Investigating Transcriptomic Heterogeneity at the Earliest Stages of Tumorigenesis
The genetic events which drive early cancer cell transformation in lung adenocarcinoma (LUAD) are well defined. In recent years, murine models of LUAD initiated by oncogenic Kras and loss of Trp53 have demonstrated that later stages of tumor progression are driven not by the acquisition of new mutations, but rather by epigenetic cell-state transitions. In fact, epigenetic heterogeneity exists within cancer cells of the same genetic background, both within and between tumors. Our lab recently developed an organoid model of alveolar type II (AT2) cells, the major cell of origin in LUAD, and we are leveraging this system to probe this epigenetic heterogeneity at the earliest stages of tumorigenesis. Specifically, we use sophisticated barcoding and next-generation sequencing technologies to track the fate of ex vivo transformed AT2 cells under external pressures and following transplantation. Our work aims to understand how transcriptomic heterogeneity within the AT2 compartment drives tumor phenotypes throughout cancer evolution and progression.
Mutation Order and Cancer Development
This project explores how the sequence in which genetic mutations occur impacts tumor development, focusing on lung cancer. Using advanced prime editing tools, we introduce common oncogenic mutations in specific orders within mouse lung AT2 organoids and in vivo models. By controlling when and where mutations are introduced, we investigate how mutational trajectories influence tumor characteristics. The study challenges the longstanding assumption that only the accumulation, not the order, of mutations matters, revealing the importance of mutation sequence in cancer biology.
Investigating the Role of Alveolar Niche Factors in Tumor Progression of Kras-mutant Lung Adenocarcinoma
Alveolar type ii (AT2) cells are an important cell of origin in lung adenocarcinoma. Using next-generation AT2 organoid models, we are investigating how alveolar niche factor availability can skew tumor progression. We can use these organoid models to understand and probe the earliest stages of tumor progression. In this way, we can ask whether or not all early tumor cells have the ability to access late-stage cell-states and/or survive in altered microenvironments. We are also investigating the genes that are important in epigenomic reprogramming of transformed cells throughout tumor progression.
In the Jacks lab, we are interested in investigating how lung tumors of hybrid cellular identities are innately resistant to targeted therapies despite being driven by the same genetic alterations as tumors of homogeneous cellular identities that tend to be significantly more sensitive to these same treatments. Lung tumors of hybrid cellular identities contain cancer cells that are highly plastic and poorly differentiated that can likely activate multiple molecular programs to survive. One way cancer cells can differentially regulate cellular processes is through altered gene expression. Therefore, we aim to define the clonality, transcriptome and epigenome of lung cancer cells from tumors of hybrid cellular identities to define differences in gene expression between these different types of lung tumors.
Our research focuses on understanding how lung adenocarcinoma develops resistance to targeted therapies through lineage plasticity, specifically via squamous transdifferentiation. Unlike resistance driven by secondary genetic mutations, this mechanism allows cancer cells to adopt a new lineage identity, enabling them to survive KRAS inhibition. We use organoid and mouse models to dissect the cellular programs and signaling pathways that accompany this transition, with the goal of identifying key regulators and potential therapeutic vulnerabilities. By elucidating the mechanisms underlying squamous transdifferentiatipon, our work aims to inform strategies that can prevent or overcome drug resistance in KRAS-driven lung cancer.
Lung Cancer Immunology Projects (click on the titles for more information)
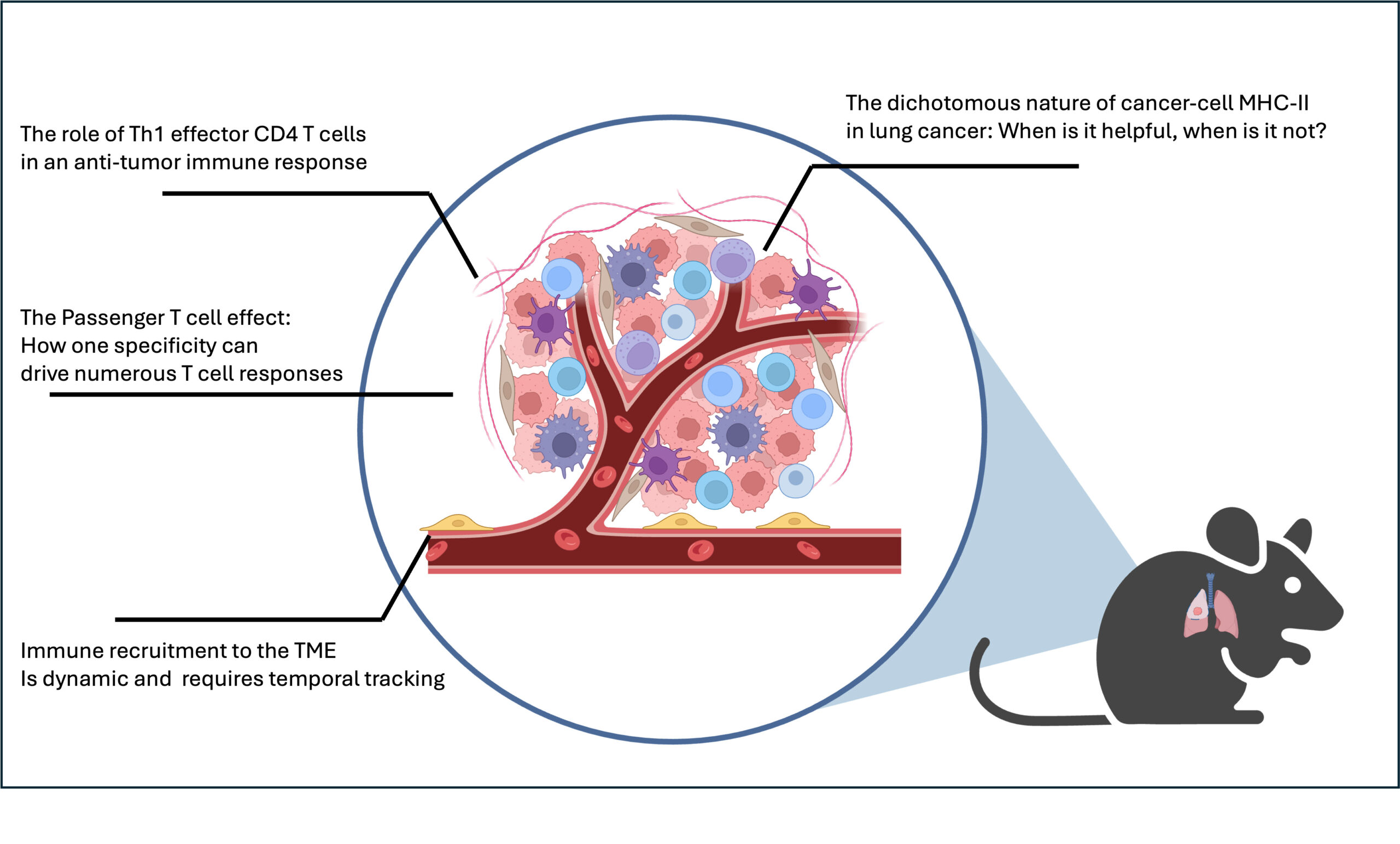
Created by Sean-Luc Shanahan using BioRender
Role of Neoantigen-specific CD4+ T cells in Lung Cancer Development
Cancer mutations can lead to the expression of novel tumor-specific antigens (termed “neoantigens”) that can be targeted by a subset of immune cells called T cells. Cytotoxic CD8+ T cells recognize MHC-I-restricted neoantigens presented on cancer cells to directly kill such cells and mediate anti-tumor immunity. CD4+ T cells recognize MHC-II-restricted antigens on antigen-presenting cells (APCs) to indirectly orchestrate immune responses, but their role in tumor-directed immunity remains incompletely understood. Two dichotomous subsets of CD4+ T cells exist: conventional CD4+ T cells (Tconv) produce cytokines and alter the phenotypes of neighboring cells to promote inflammation, while Foxp3+ CD4+ regulatory T cells (Treg) act to suppress ongoing immune responses and are thought to be a major barrier to anti-tumor immunity. It is currently unclear how neoantigen-specific CD4+ T cells impact tumor development and coordinate associated immune responses throughout disease progression. Our laboratory has developed an autochthonous mouse model of lung adenocarcinoma (termed “KP mice”) in which tumors expressing MHC-II-restricted neoantigens can be initiated at a defined timepoint and studied over time. We unexpectedly observe a higher tumor burden in mice when a single MHC-II-restricted neoantigen is expressed, suggesting that neoantigen-specific CD4+ T cells promote tumorigenesis.
Our objective is to determine how neoantigen-specific CD4+ T cells promote lung cancer tumorigenesis. Leveraging our KP model, we will establish a system in which the development and progression of tumors expressing or lacking and MHC-II-restricted neoantigen, and their accompanying immune responses, can be studied in a single unified setting. It is our central hypothesis that neoantigen-specific CD4+ T cells establish an environment early in tumor development that promotes disease progression. We aim to define the temporal windows, anatomical locations, and mechanisms by which neoantigen-specific CD4+ T cell responses promote tumorigenesis.
CD4+ T cells recognize their cognate antigen presented on MHC-II to help coordinate and augment concurrent immune responses. Classically, professional immune cells were thought to be the only cell populations capable of presenting MHC-II complexes to CD4+ T cells and orchestrating their responses. Recently, cancer cells in lung adenocarcinoma have been discovered to also express MHC-II, evident in human cancer biopsy samples and our lab’s autochthonous murine lung cancer models. Notably, other cancer types such as melanoma and colorectal cancers also express MHC-II and thus have the potential to directly engage with CD4+ T cells. We are interested in understanding the roles of such cancer cell MHC-II expression in shaping CD4+ T cell responses and resulting tumor progression in lung adenocarcinoma.
These studies will yield unprecedented insight into naturally-occurring T cell responses that are clinically relevant to lung cancer progression and are current targets for emerging therapeutics. By uncovering the role of neoantigen-specific CD4+ T cells throughout cancer progression, we expect to identify temporal windows for effective CD4+ T cell-directed therapies and reveal cell populations that can be synergistically targeted to enhance current and emerging immunotherapies.
Role of Nonspecific Passenger T cells in Cancer-associated Immunity and Tumor Progression
T cells are key mediators of anti-tumor immune responses, and their presence in tumors can be a predictor of patient response to immunotherapy. However, populations of immunosuppressive T cells, such as Foxp3+ regulatory T cells, exist alongside anti-tumorigenic T cells and are a major barrier to durable tumor control and elimination. Given this dichotomous nature of T cell responses, we must pioneer new approaches that leverage the untapped potential of cancer-fighting T cells while repressing those that shut down immune responses. CD8+ and CD4+ T cells use T cell receptors (TCRs) to recognize peptide antigens presented on the molecules MHC-I and MHC-II, respectively. In tumors, T cells can mount targeted immune responses against cancer cells, where mutations lead to the expression of novel tumor-specific antigens (termed “neoantigens”) that are recognized by neoantigen-specific T cells. A substantial effort has been made to leverage the ability of neoantigen-specific T cells to directly kill cancer cells or propagate tumor-associated immune responses. However, neoantigen-specific T cells eventually lose their anti-tumor potential via T cell dysfunction and exhaustion, as tumors evolve to downregulate the neoantigen, or upon establishment of a suppressive environment within the tumor.
Emerging evidence suggests that T cells not specific for neoantigens play an unappreciated role in tumor-associated immune responses and immunotherapies. In mouse models of cancer, the expression of a defined neoantigen by cancer cells results in robust T cell infiltration of tumors, yet the vast majority of these cells have no detectable reactivity to the neoantigen and include both pathogenic and immunosuppressive subsets. These nonspecific “Passenger” T cells may react to a variety of antigens, including those derived from pathogens, expressed during embryonic or tissue development, or unmutated self-proteins that are normally not targeted by the immune system. Curiously, emerging models of chronic autoimmune diseases describe a similar phenomenon, whereby the effective attack of a single self-antigen results in tissue-specific infiltration of nonspecific Passenger T cells thought to sustain and exacerbate disease by targeting other self-antigens. A critical outstanding question lies in understanding how neoantigen-specific T cells orchestrate the enrichment of Passenger T cells in tumors, and whether such cells can be reprogrammed to mount durable, destructive tissue-specific immunity as observed in autoimmune disease.
We aim to elucidate the identity and function of Passenger T cells throughout tumor progression and understand their relationship to neoantigen-specific T cells. This approach is a departure from historical strategies that focus on the biology of neoantigen-specific T cells in isolation by instead aiming to demystify the accompanying T cells and their impact on cancer progression. We aim to define the landscape of Passenger T cells throughout progression to identify unique and shared populations driven by CD8+ and CD4+ neoantigen-specific T cells, determine how Passenger T cells impact tumor control or progression, and investigate the mechanisms driving the enrichment of Passenger T cells.
These studies promise to expand the breadth of T cells contributing to productive and durable anti-tumor immunity and lay the foundation for next-generation therapies to treat both cancer and autoimmune disease.
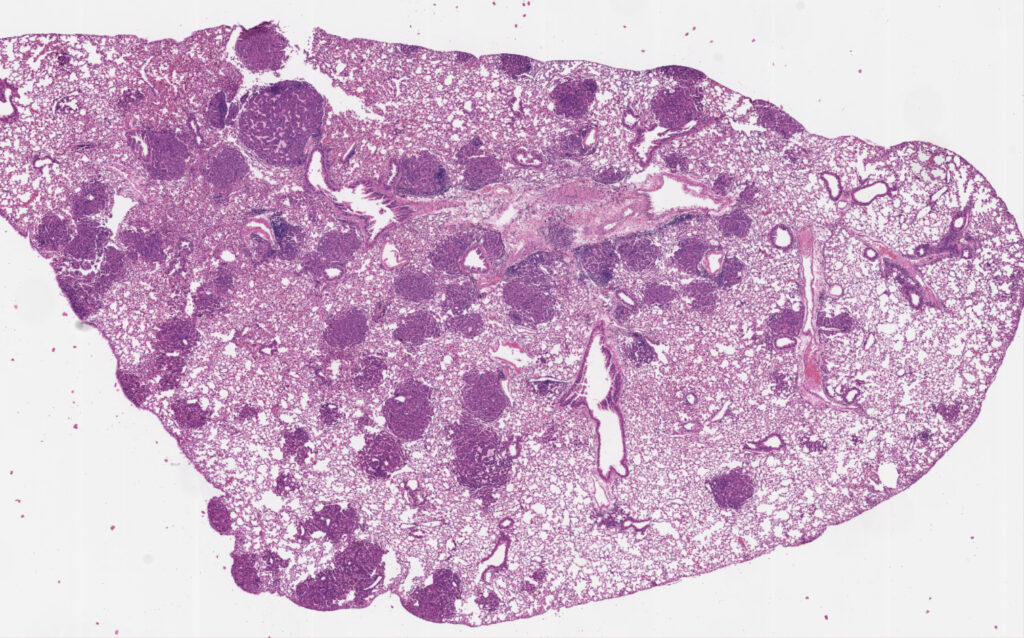
Pictured: H/E stained section of a mouse lung containing several tumor nodules. Image was generated by David Klawon.
MHC-II antigen Presentation on Cancer Cells Improves CD4+ T cell Immunity and Vaccine Efficacy
CD4+ T cells can be either protective or pathogenic in cancer, but how they interact with cancer cells to produce these effects remains poorly understood. Previously, we observed that expression of a CD8+ T cell antigen (SIINFEKL) initially delays tumor growth, but cancer cells eventually persist while still expressing the antigen. We developed a new iteration of this autochthonous platform to introduce CD4+ T cell antigens alongside CD8+ T cell antigens in a mouse lung cancer model. Strikingly, we found that tumor-specific CD4+ T cells potentiate CD8+ T-cell control of tumor progression, resulting in either clearance of cancer cells or immunoediting of the tumor antigens. Unlike previous studies emphasizing intratumoral interactions among dendritic cells, CD4+ T cells, and CD8+ T cells, we found that CD4+ T cell function and tumor control depend on direct antigen presentation by cancer cells via major histocompatibility complex class II (MHC-II). This direct interaction between cancer cells and CD4+ T cells led to increased immune infiltration, inflammatory remodeling, and sustained CD8+ T cell effector functions within tumors. Building on this, we demonstrate that tumor vaccination targeting both CD4+ and CD8+ T cell neoantigens significantly reduces tumor burden and requires cancer cell MHC-II presentation. These finds reveal an underappreciated mechanism of CD4+ T cell “help” and suggest a therapeutic approach targeting cancer cell presentation of MHC class II antigens.
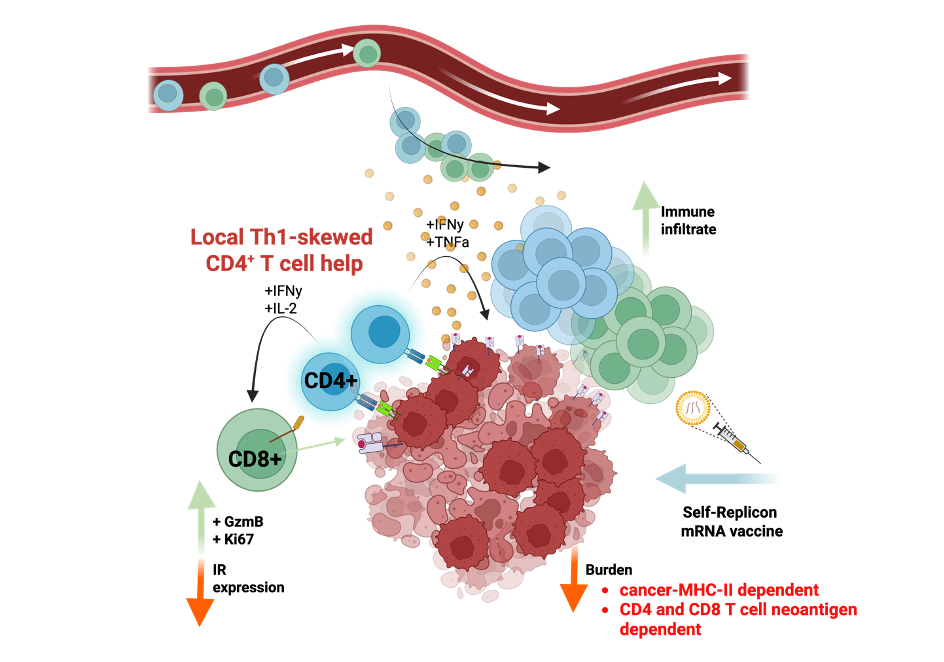
T Cell infiltration supports early lung adenocarcinoma formation through engagement with cancer MHC-II Efficacy
The immune system is tasked with maintaining tissue homeostasis in the body. The immune system responds to disruptions in homeostasis – such as an infection, tissue damage, or tumor development – through a balance of clearing potential pathogens and abnormal cells and aiding in the regeneration of healthy tissue. To successfully respond to disruptions in tissue homeostasis, immune cells constantly survey tissues for abnormalities. Among them, CD4+ T cells and their specialized subsets coordinate and tune such responses to the specific nature of each disruption. Historically, CD4+ T cell surveillance was thought to be restricted to CD4+ T cells only engaging with their cognate antigens when presented on MHC-II complexes by professional antigen presenting immune cells (APCs). Recently it is becoming evident that other non-immune cells, particularly populations of cells that exhibit stem-like properties, have the capacity to express MHC-II and engage CD4+ T cells. The roles of such cells expressing MHC-II influencing CD4+ T cells and subsequent balance between tissue regeneration and destruction remain limited. Interestingly, multiple cancer types – such as lung, melanoma, and colorectal – express MHC-II at early stages of tumor development. Preliminary data from our lab demonstrate a cancer cell dependency on T cell infiltration during early tumor development. We seek to understand how cancer cell MHC-II expression influences infiltrating CD4+ T cells during early stages of lung adenocarcinoma and how this interaction shapes the balance between destructive and regenerative immune programs.
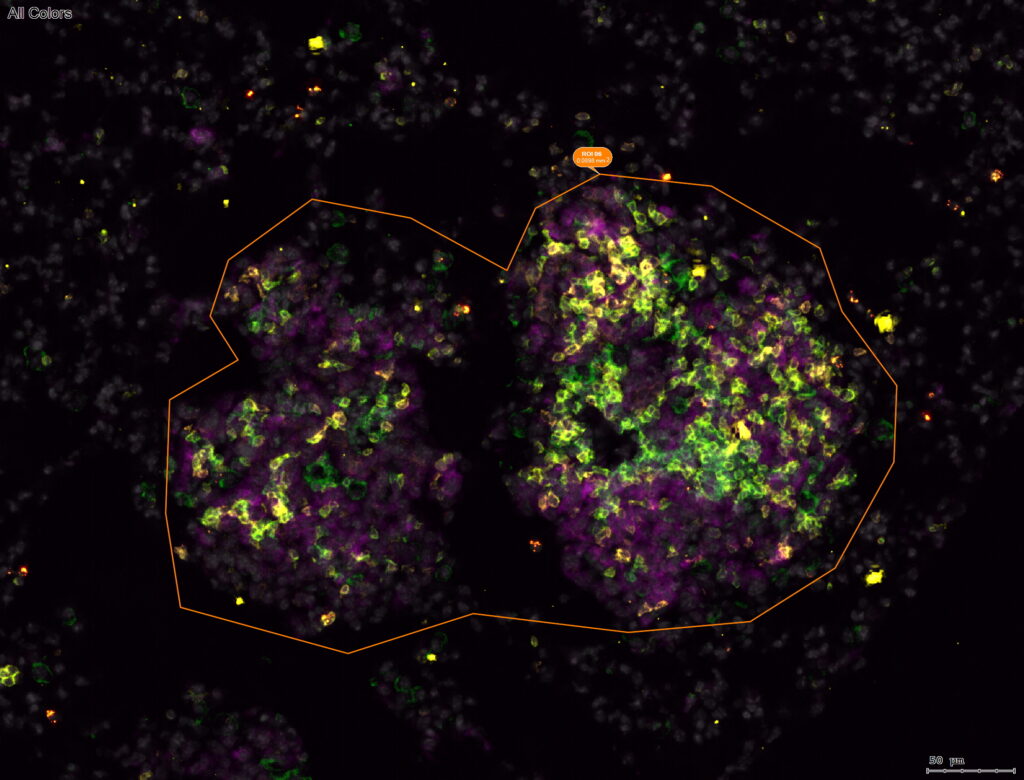

Image was generated by Jacob Kassama.